Перевод А. Белоусов, sarcoidosis@yandex.ru
Глава 4
Иммунология и патофизиология
G. Semenzato, M. Bortoli, E. Brunetta, C. Agostini
Dept of Clinical and Experimental Medicine, Padua University School of Medicine, Padua, Italy.
Correspondence: G. Semenzato, Universita` di Padova, Dipartimento di Medicina Clinica e Sperimentale, Immunologia Clinica, Via Giustiniani 2, 35128 Padova, Italy. Fax: 39 0498754179; E-mail: g.semenzato@unipd.it
C тех пор как стало известно, что иммунорегуляторные механизмы, которые участвуют в развитии саркоидной гранулемы, могут модулировать патогенетические события, ведущие к развитию гранулем при других гранулематозных болезнях, саркоидоз стал рассматривается как иммунная гранулематозная болезнь [1]. Существенный прогресс был достигнут в понимании общих иммунологических и молекулярных аспектов механизмов, ведущих к формированию гранулемы и развитию фиброза при саркоидозе. В частности, саркоидная гранулема рассматривается как последствие нарушения иммунологического ответа против неизвестного антигена, который персистирует на участках воспаления, возможно из-за его низкой растворимости и слабой деградации. Действительно, хотя реакции гиперчувствительности обычно разрешаются, баланс между событиями, которые приводят к разрешению или продлению воспалительного ответа, у пациентов с хроническим саркоидозом может быть нарушен. Персистирование этиологических агентов и/или нарушения механизмов удаления воспалительных клеток и продуктов их секреции, в конечном счете приводит к продолжительному воспалительному ответу. В результате происходит локальное производство цитокинов с провоспалительными и деструктивными биологическими свойствами. При участии этих цитокинов происходит необратимое ремоделирование ткани легкого, прогрессирующее формирование легочных гранулем и, у некоторых индивидуумов, развитие необратимого легочного фиброза.
Цель настоящей главы состоит в том, чтобы дать краткий обзор доступных в настоящее время знаний о механизмах, ведущих к воспалительным процессам, которые происходят на участках активности саркоидоза. Также приведено детальное описание клеточных взаимодействий, которые управляют динамикой формирования гранулемы.
Роль инфильтрации воспалительными клетками в формировании саркоидных гранулем
Механизмы, ведущие к T-клеточной инфильтрации на участках активности болезни
Инфильтрация активизированными CD4+ T-клетками является иммунологическим признаком саркоидоза (Таблица 1). Хотя паренхима легкого в норме содержит лишь отдельные лимфоидные элементы, при саркоидозе наблюдается поразительная компартментализация популяций лимфоцитов в воздушных пространствах и интерстиции. В жидкоcти бpонxоальвеоляpного лаважа (ЖБАЛ) пациентов с активным легочным саркоидозом, может быть обнаружено примерно 25*106 T-клеток, наиболее вероятно в ответ на неизвестный экзогенный или эндогенный антиген(ы). Саркоидные T-клетки в основном имеют фенотип CD4+, CD45R0, коэкспрессирующие ab T-клеточный рецептор (ТКР), производящие главным образом интерферон гамма (IFN-g) или интерлейкин-2 (IL-2) и, таким образом, принадлежащие к семейству T-хелперов типа 1 (Th1). Значительное накопление CD4+ лимфоцитов может наблюдаться во всех тканях, пораженных саркоидным воспалительным процессом [2, 3], с чрезвычайно высоким отношением CD4:CD8 (обычно >10).
Таблица 1. Иммунологические особенности, которые наблюдаются у пациентов с саркоидозом
| Т-клетки | Инфильтрация CD4+ клетками, секретирующих Th1 цитокины с существенным увеличением отношения CD4:CD8 на участках активности болезни. |
| Экспансия T-клеток, несущих ограниченный репертуар Vb и Va ТКР в пораженных тканях. Этот паттерн совместим с олигоклональностью ТКР. | |
| Увеличенная экспрессия рецепторов и молекул - членов суперсемейства ФНО, которые регулируют время жизни и смерти саркоидных T-клеток. Это говорит, что нарушение механизма апоптоза T-клеток может объяснять отсутствие апоптоза в гранулеме и персистирование воспаления. | |
| Увеличенная экспрессия рецепторов хемокинов, участвующих в рекрутировании активизированных Th1 клеток (CXCR3, CCR6, CXCR6). | |
| Макрофаги | Накопление макрофагов с увеличенной экспрессией маркеров активации, молекул адгезии (CD49a, CD54, CD102) и костимулирующих молекул (CD80 и CD86, CD40 и CD72). |
| Легочные макрофаги имеют увеличенную антигенпрезентирующую способность. | |
| Производство цитокинов (IL-1, IL-6, IL-15, ФНО-a, GM-CSF) макрофагами, приводящее к повреждению ткани и формированию гранулем. | |
| Производство хемокинов, которые стимулируют формирование гранулем (CCL5, CCL20, CXCL9, CXCL10, CXCL16) | |
| Производство фиброгенетических цитокинов (TGF-b, PDGF, IGF-I), вовлеченных в прогрессирование болезни к фиброзу. |
Сокращения: Th: клетки T-хелпер; V: вариабельный регион; ТКР: Т-клеточный рецептор; ФНО: фактор некроза опухоли; CXCR: рецептор CXC хемокина; CCR: рецептор CC хемокина; IL: интерлейкин; GM-CSF: гранулоцитомакрофагальный колониестимулирующий фактор; CCL: лиганд CC хемокина; CXCL: лиганд CXC хемокина; TGF: трансформирующий фактора роста; PDGF: фактор роста тромбоцитов; IGF-I: инсулино-подобный фактор роста.
T-клетки на участках активности болезни обладают ограниченным репертуаром вариабельного региона Vb и Va Т-клеточных рецепторов (ТКР), что совместимо с олигоклональностью ТКР. Ограниченность репертуара ТКР при саркоидозе может быть объяснена с помощью различных механизмов. Одна из гипотез состоит в том, что предполагаемый антиген(ы) приводит к экспансии олигоклональных T-клеток, имеющих специфический Va и Vb регион. Кроме того, in situ производство цитокинов также вероятно играет роль в этом явлении. При стимуляции IL-2, T-клетки пациентов с саркоидозом демонстрируют селективную экспансию специфических субпопуляций, экспрессирующих Vb. Секвенирование скользящих (junctional) регионов показало, что стимулированные IL-2 T-клетки являются поразительно олигоклональными и происходят от клонов T-клеток, уже селективно сгруппированных in vivo. Таким образом, антиген(ы), который вызывает развитие гранулематозных повреждений, стимулирует прогрессирующую аккумуляцию и активацию ограниченного числа клонов Th1 клеток. Когда воспалительными Th1 клетками поражено достаточное количество ткани, появляются клинические признаки активности болезни, которые ощущаются индивидуумом, например, как одышка, когда поражен респираторный тракт.
Рис. 1. После опознавания неизвестного антигена, вызывающего саркоидоз, альвеолярные макрофаги (AM) производят IL-12 и IL-18. Оба цитокина участвуют в активации макрофагов. Это событие стимулирует производство макрофагальных хемокинов (CXC) и цитокинов (IL-15), участвующих в рекрутировании Th1 клеток. В свою очередь, рекрутированные Th1 клетки производят IFN-g, который стимулирует дальнейшую аутокринную петлю. Результат - развитие T-клеточного альвеолита.
Гомеостаз T-клеток регулируется растворимыми факторами и мембранными рецепторами, которые активизируют пролиферативные и апоптические процессы (рис. 1). С начала 1980-х годов было известно, что in situ пролиферация Th1 клеток является механизмом, ответственным за накопление CD4 клеток в саркоидных тканях (см. позже). Множество данных также говорит, что связывание рецепторов передачи сигнала клеточной смерти или модуляция апоптоза T-клеток может иметь нежелательные патогенетические эффекты у пациентов с прогрессирующим саркоидозом. Как было недавно показано, у саркоидных T-лимфоцитов наблюдается резистентность к апоптозу [4], которая может вносить вклад в накопление воспалительных клеток в легких, в персистирование воспаления, в развитие и стабильность гранулем. Интересно, что процент регулирующих T-клеток (Tr), определенных как CD4+ и CD25+(bright) лимфоциты, увеличен в легких пациентов с активной болезнью, которые имеют спонтанное клиническое разрешение болезни, предполагая, что увеличенное число Tr при активном саркоидозе может способствовать уменьшению клеточного иммунного ответа [5].
Фактор некроза опухоли альфа (ФНО-a) также может играть противоречивую роль в модуляции активности саркоидных T-клеток на участках воспаления. Имеются данные, предполагающие, что хроническая экспрессия ФНО-a и IFN-g производит состояние персистирования и прогресса воспалительных событий у пациентов с хроническим саркоидозом. В некоторых обстоятельствах, изменение баланса рецептор/лиганд ФНО, ведет к хроническому рекрутированию воспалительных клеток, которое в уже воспаленных тканях, производит новые гранулематозные структуры. Наоборот, имеются данные, указывающие, что ФНО-a необходим для рекрутирования Th1 клеток, формирования гранулемы, уничтожения антигена, и, таким образом, для разрешения гранулематозной болезни [6]. Вероятно, возможны оба эффекта. ФНО-a может быть необходим или может иметь небольшой эффект на механизмы апоптоза в пределах гранулематозных структур, в зависимости от комбинации генетических факторов, предыдущего воздействия факторов окружающей среды и локальных изменений имунокомпетентности.
P21, член семейства Cip/Kip, регулирует клеточную жизнь и смерть, что подразумевает, что он является главным регулятором судьбы клетки. Активация макрофагов in vitro IFN-g или их адгезия к внеклеточному матриксу ведет к подавлению апоптоза [7, 8]; медиатором этого эффекта является экспрессия p21 (Waf1). В недавнем исследовании, высокие уровни p21 были обнаружены при легочном саркоидозе и вероятно, что гиперэкспрессия p21 могла бы объяснить отсутствие апоптоза в гранулеме и персистирование воспаления [9].
Другая система, вовлеченная в регулирование T-клеточных воспалительных процессов - система Fas/Fas лиганд (FasL). Fas, который сильно экспрессирован при высоких уровнях хронической стимуляции T-клеток, ограничивает экспансию антиген-реактивных T-клеток после лигирования со специфическим лигандом, принадлежащем суперсемейству лигандов ФНО (FasL), таким образом предотвращая чрезмерное накопление активизированных антигеном лимфоцитов [10]. Обе эти системы были оценены при саркоидозе [11]. Молекулы Fas у саркоидных T-лимфоцитов имеют больший уровень экспрессии, чем у нормальных субпопуляций T-клеток [12]. Кроме того, высокие концентрации растворимой формы FasL, которая связана с уменьшением цитотоксичности, были обнаружены в ЖБАЛ и сыворотке пациентов с саркоидозом, но не у здоровых индивидуумов [13].
Запрограммированная смерть T-клеток также ингибируется онкогенами. Онкоген BCL2, кодируемый геном Bcl-2, принадлежит к семейству апоптоз-регулирующих белков, которые могут быть антагонистами или агонистами клеточной смерти. Увеличенная экспрессия некоторых членов семейства (например, BCL2 и гена кодирующего Bcl-2-like 1 (BCLXL)), защищает лимфоидные клетки от запрограммированной клеточной смерти в отсутствие некоторых факторов роста, например IL-2, принимая во внимание, что увеличенная экспрессия других членов семейства (например, генов кодирующих апоптические белки BAD, BAX и BID), блокирует поступающие сигналы от рецепторов цитокинов и стимулирует апоптоз. Подобно Fas, BCL2 сильно экспрессирован T-лимфоцитами, окружающими гранулематозные повреждения пациентов с саркоидозом [14]. Было показано, что множество связанных с апоптозом факторов, включая факторы роста и семейство генов BCL2, у пациентов с саркоидозом сверхэкспрессированы, что совместимо с профилем увеличенной выживаемости клеток [15]. Кроме того, у пациентов с прогрессирующей болезнью наблюдается увеличеная экспрессия ядерного фактора NF-kappaB и ослабление регуляции ингибиторами апоптоза [15].
Также было показано, что взаимодействие с фибробластами может ингибировать апоптоз активизированных цитокинами T-клеток селективным эффектом BCLXL: медиатором этого явления вероятно являются растворимые факторы, произведенные фибробластами, которые снижают синтез глутатиона и поддерживают высокие уровни BCLXL, что помогает поддерживать гранулематозный процесс. Поэтому, возможно, что увеличенная экспрессия этого ингибитора апоптоза может предотвращать гибель активизированных T-клеток.
Влияние произведенных Th1 клетками молекул на воспалительные события при саркоидозе
Данные о паттерне производства лимфокинов в течение саркоидоза могут быть получены в контексте парадигмы Th1/Th2. Идея состоит в том, что изменение баланса Th1/Th2 играет важную роль в детерминировании организации саркоидной гранулемы и прогрессе болезни. Цитокины, производимые T-клетками и макрофагами, которые регулируют баланс Th1/Th2, обсуждены в следующей секции.
Th1 цитокины. IFN-g - ключевой фактор, который вызывает все воспалительные процессы при саркоидозе (рис. 1). IFN-g экспрессируется Th1 клетками, инфильтрирующими пораженные ткани и способствует развитию типичной реакции гиперчувствительности и ингибирует фиброгенетические процессы. Важную роль этого цитокина подтверждает наличие высоких уровней IFN-g в ЖБАЛ пациентов с активным саркоидозом [16].
Посредством своих плейотропных эффектов на производство цитокинов, IFN-g увеличивает экспрессию костимулирующих молекул на акцессоpных клетках, включая CD80 и CD86 [17]. IFN-g также имеет важный антифиброзный эффект, так как способен ингибировать пролиферацию эндотелиальных клеток и синтез коллагена фибробластами. Как сообщалось ранее, также имеется существенная корреляция между увеличенной экспрессией IFN-g и экспрессией ингибитора cdk p21/Waf1 (анти-апоптическая молекула) в легком при саркоидозе [9], предполагая, что высокие уровни p21, индуцированные IFN-g, могут объяснить отсутствие апоптоза в гранулеме и персистирование воспаления.
Однако, с помощью индуцирования не ERL хемокинов (MIG/CXCL9, IP-10/CXCL10, ITAC/CXCL11), IFN-g также играет главную роль в рекрутировании и активации саркоидных CXCR3+ T-клеток в воспаленных тканях (рис. 1). Действительно, имеется высокое число CD4+ T-клеток, экспрессирующих CXCR3, CCR5, IL-12R и IL-18R в легком пациентов с саркоидозом, принимая во внимание, что рецепторы Th2 хемокинов CXCR4 и CCR4 экспрессированы у небольшого процента легочных саркоидных CD4+ T-клеток [18].
IL-2 активно производится на участках активности болезни при саркоидозе, где действует как местный фактор роста T-лимфоцитов, инфильтрирующих пораженные ткани [19-21]. Было продемонстрировано присутствие сайтов сцепления с IL-2 у легочных фибробластов человека. Сцепление IL-2 с фибробластами ведет к увеличению экспрессии гена, кодирующего белки MCP-1 и CCL2, которые вовлечены в фиброз посредством регулирования генерации профибротических цитокинов и матрикса. Таким образом, IL-2 может интегрировать участие фибробластов и саркоидных макрофагов в скоординированном ответе, инициированном Th1 лимфоцитами на участках активности болезни.
Th2 цитокины. Переключение на Th2 паттерн может происходить у пациентов с прогрессирующим саркоидозом. У этих пациентов, T-клетки производят Th2 цитокины, включая IL-4, который является кофактором пролиферации многих линий клеток, включая фибробласты [22-24]. Активизированные Th2 клетки также являются источником IL-10, который имеет противовоспалительные и иммунорегуляторные свойства: он ингибирует провоспалительные цитокины и производство хемокинов в дополнение к блокированию ответа T-клеток на специфические антигены. Было предположено, что местная секреция IL-10 может быть модуляционным механизмом, вовлеченным в спонтанное разрешение альвеолита при саркоидозе [25]. Недавно полученные данные говорят, что низкое производство IL-22, члена семейства IFN типа 1, которое также включает IL-10, может участвовать в патогенезе саркоидоза [26]. IL-22 потенциально может взаимодействовать с IL-10, поскольку наблюдается сцепление цепи IL-10R2C и рецептора IL-22R1. Точная роль IL-22 в патогенезе саркоидоза остается неустановленной. Наконец, IL-13, Th2 цитокин, который экспрессирован CD4+ T-клетками и подавляет ФНО-a, активно производится в легких некоторых пациентов с саркоидозом [27]. Однако, клеточным источником этого про-Th2 цитокина при саркоидозе скорее являются альвеолярные макрофаги, чем Th2 клетки. В любом случае, увеличенное производство IL-13 могло бы иметь противовоспалительный эффект на окружающую микросреду, влияя на производство TNF-a.
Молекулы, произведенные моноцитами/макрофагами
Взаимодействие между IFN-g и его рецепторами активизирует макрофаги в ранней стадии саркоидного воспалительного процесса (рис. 1). Это состояние активации саркоидных макрофагов характеризуется увеличенной секрецией иммуномодуляционных молекул (Таблица 1), как описано в следующей секции.
Провоспалительные цитокины. Саркоидные макрофаги способны к производству обнаружимых количеств IL-1, который регулирует развитие альвеолярного воспаления при саркоидозе. Кроме того, IL-1 per se может стимулировать формирование гранулемы и развитие фиброза, стимулируя пролиферацию фибробластов и увеличивая производство коллагена. Как обсуждалось ранее, ФНО-a - другой провоспалительный цитокин, активно производимый саркоидными макрофагами. Он играет критическую роль в повреждении легкого и регулировании роста фибробластов посредством индукции IL-6. Кроме того, ФНО-a стимулирует и регулирует синтез других лимфокинов (IL-1, GM-CSF, фактор активации тромбоцитов и IL-6) и увеличивает производство простагландинов (простагландин E2). Имеются данные, предполагающие, что хроническая экспрессия ФНО-a и IFN-g устанавливает состояние персистирования и прогресса воспалительных событий и повреждений ткани при саркоидозе [12, 28- 30]. Это предполагает важность анти-ФНО стратегий для лечения саркоидоза.
Цитокины, вовлеченные в регулирование баланса Th1/Th2 клеток. IL-12, главная молекула, вовлеченная в инициирование Th1 иммунного ответа, была интенсивно изучена. Было ясно показано ее участие в развитии гранулем в легком, включая саркоидоз [31-34]. IL-12 стимулирует пролиферацию активизированных саркоидных T-клеток. В синергизме с IL-15, IL-12 стимулирует взаимодействие между активизированными T-клетками и саркоидными макрофагами и стимулирует экспрессию рецепторов хемокинов Th1 клетками (рис. 1). Этот цитокин действует с помощью взаимодействия со специфическими рецепторами (IL-12Rb), экспрессированных лимфоцитами, которые накапливаются на участках активности болезни [35].
IL-27, недавно описанный член семейства IL-12, также вовлечен в патогенез формирования гранулемы [36]. Этот цитокин действует в синергизме с IL-12 и состоит из двух субъединиц, p28 и EBI3. Иммуногистохимические исследования гранулематозных тканей показали, что коэкспрессия EBI3 и p28 может быть обнаружена у эпителиоидных и многоядерных гигантских клеток саркоидных гранулем. Кроме того, тканевые макрофаги, эндотелиальные и плазматические клетки коэкспрессируют EBI3 и p28. Эти данные говорят, что IL-27 может играть определенную роль в патогенезе формирования гранулемы, хотя молекулярные механизмы, посредством которых IL-27 участвует в формировании центрального макрофагального ядра гранулемы, остаются неизвестными.
Другим про-Th1 цитокином, который действует совместно с IL-12, является IL-18 (рис. 1). Произведенный главным образом моноцитами и макрофагами, IL-18 стимулирует экспрессию IFN-g и GM-CSF, но ингибирует производство IL-10. IL-18 и IL-12 действуют в синергизме на саркоидные Th1 клетки [37-40]. Однако, было показано, что IL-18, посредством активации белка активации 1 и NF-kappaB, ведет к увеличенной экспрессии гена IL-2 при саркоидозе [37].
В подтверждение локальных эффектов IL-18 было показано, что пациенты с активной болезнью демонстрируют гиперэкспрессию факторов транскрипции семейства NF-kappaB [41]. Недавно полученные результаты говорят, что местная гиперэкспрессия этих регуляторов немедленного транскрипционного ответа при саркоидозе может до некоторой степени регулироваться генетическими факторами [42, 43]. Некоторые молекулярные механизмы, которые стимулируют экспрессию NF-kappaB у саркоидных макрофагов, начинают проясняться. Рецептор PPAR-g, имеет важное влияние на модуляцию Th1 иммунного ответа, частично уменьшая производство воспалительных цитокинов. Саркоидные макрофаги демонстрируют активацию NF-kappaB и дефицит PPAR-g, предполагая, что недостаточная активность PPAR-g вносит вклад в персистирование воспаления при легочном саркоидозе, вследствие неспособности подавить провоспалительные факторы транскрипции, такие как NF-kappaB [44].
Саркоидные макрофаги также производят IL-15, цитокин, который поддерживает рост, выживание и хемотаксис саркоидных T-клеток, стимулируя развитие Th1 инфильтрации [45, 46]. IL-15 также ведет себя как стимулирующий фактор при производстве других цитокинов и хемокинов (IL-17, CXCL8/IL-8, CCL2/MCP-1, GM-CSF, IFN-g и ФНО-a) и экспрессии молекул, определяющих антигенпрезентирующую способность резидентных акцессорных клеток (CD80/CD86). Кроме того, обнаружение, что IL-15 уменьшает апоптоз легочных T-клеток, указывают на IL-15 как на возможный ингибитор физиологических стимулов апоптоза, который приводит к персистированию воспалительных процессов на участках активности болезни.
Колониестимулирующие факторы. Колониестимулирующие факторы, и в частности GM-CSF, активно производятся саркоидными макрофагами. GM-CSF способен стимулировать рост и дифференцирование саркоидных макрофагов, стимулируя развитие ядра саркоидной гранулемы [47, 48]. Кроме того, GM-CSF модулирует производство цитокинов и увеличивает антигенпрезентирующую способность саркоидных макрофагов [49].
Хемокины
Траффик и накопление иммунокомпетентных клеток - необходимые компоненты в патофизиологии воспалительных процессов, имеющих место в легком пациентов с саркоидозом. Множество данных говорит, что большинство этих событий регулируется хемокинами, которые сверхэкспрессированы в легком пациентов с саркоидозом (Таблица 2).
Таблица 2. Хемокины и рецепторы хемокинов, вовлеченные в патогенез саркоидоза
| Хемокин | Лиганд | Рецептор |
| CC хемокины | ||
| CCL2 | MCP-1, MCAF | CCR2 |
| CCL3 | MIP-1, LD78a, LD78b, AT464.1, AT464.2,GOS19-1, GOS19-2 | CCR1, CCR5 |
| CCL4 | MIP-1b, AT744.1, AT744.2, Act-2, G-26, HC21, H400, LAG-1 | CCR5 |
| CCL5 | RANTES | CCR1, CCR3, CCR5 |
| CCL19 | MIP-3b/exodus-3 | CCR7 |
| CCL20 | MIP-3a/exodus-1 | CCR6 |
| CXC хемокины | ||
| CXCL8 | IL-8, MDNCF, NAP-1, NCF | CXCR1, CXCR2 |
| CXCL9 | MIG, HuMIG | CXCR3 |
| CXCL10 | IP-10 | CXCR3 |
| CXCL11 | ITAC, H174, b-R1 | CXCR3 |
| CXCL16 | SR-PSOX | CXCR6 |
Воспалительный белок моноцитов MIP-3b/CCL19, участвующий в рекрутировании T-лимфоцитов при саркоидозе, связан с прогрессом болезни и его экспрессия уменьшается препаратами, используемыми для лечения саркоидоза [50]. Однако, высокие уровни MCP-1/CCL2, MIP-1a/CCL3, MIP-1b/CCL4 и RANTES/CCL5 действуют совместно, чтобы иммобилизировать несколько субпопуляций лейкоцитов в периваскулярных очагах воспаления. MCP-1/CCL2 и RANTES/CCL5, взаимодействуя с CCR1/CCR2 или CCR1/CCR3/CCR5, соответственно, могут быть хемоаттрактантами для различных клеток, которые характеризуют различные стадии саркоидного воспалительного процесса, включая макрофаги и T-лимфоциты.
Три лимфоцит-специфических CXC хемокина, которые производятся в ответ на IFN-g (CXCL10, MIG/CXCL9 и ITAC/CXCL11) [51], играют важную роль в рекрутировании активизированных T-клеток в легочную микросреду. Саркоидные макрофаги - главный клеточный источник этих молекул; они производят большое количество CXCL10 и CXCL9, которые, взаимодействуя со специфическими рецепторами, экспрессированными Th1 клетками (CXCR3), участвуют в накоплении легочных T-лимфоцитов и вносят вклад в формирование гранулемы [52]. Активизированный бронхиальный эпителий - другой важный источник CXCL9, CXCL10 и CXCL11.
Авторы этой главы недавно оценили, действительно ли CXCR3 коэкспрессирован вместе с другими Th1 рецепторами хемокинов. Предварительные данные указывают, что два других типичных Th1 рецептора хемокинов, CCR6 и Bonzo/CXCR6, экспрессированы саркоидными Th1 клетками. В частности показано, что, принимая во внимание, что CXCR3 рано экспрессирован саркоидными T-клетками, экспрессия CXCR6 - результат длительного воздействия IFN-g и макрофагальных цитокинов, прежде всего IL-15 в легочной микросреде.
IL-8/CXCL8, хемокин, который стимулирует рекрутирование T-клеток и нейтрофилов, активно производится в дыхательных путях при саркоидозе и его производство связано с повреждением легкого. Иммунолокализация IL-8 продемонстрировала, что легочные фибробласты - основной клеточный источник IL-8, даже при том, что имеются данные, предполагающие, что макрофаги также могут производить этот хемокин. Интересно, что легочный фиброз может быть связан с увеличенным производством IL-8/CXCL8 и дисрегуляцией производства CXCL10, предполагая, что баланс хемокинов является важным фактором в регулировании локального ангиогенеза и фиброгенеза.
Все эти данные подчеркивают важную роль хемокинов в развитии саркоидного воспаления. Также вероятно, что взаимодействия клетка-клетка и клетка-матрикс модулируют местную экспрессию хемокинов, способствуя прогрессу воспалительных повреждений к фиброзу. Как было недавно отмечено [29], различными фармацевтическими компаниями разрабатывается множество антагонистов рецепторов хемокинов. Специфические антагонисты хемокинов приближаются к первым клиническим исследованиям. Терапевтическое использование молекул, селективных для рецепторов хемокинов, возможно имеет большой потенциал для лечения саркоидоза и других диффузных болезней легкого.
Взаимодействие макрофагов и T-клеток - ключевой фактор в формировании гранулемы
Ядро типичной саркоидной гранулемы состоит из множества моноцитов / макрофагов в различных состояниях активации и дифференцирования, эпителиоидных клеток и многоядерных гигантских клеток. Факторы хемотаксиса и факторы активации лейкоцитов, которые активно секретируются в тканях, пораженных саркоидозом, способны к рекрутированию моноцитов из крови, формируя центральную структуру гранулемы.
Гистопатологические данные продемонстрировали присутствие макрофагов и антигенпрезентирующих клеток (APCs) в T-клеточных областях гранулемы [53]. Эти клетки, даже находясь в составе зрелых компонентов гранулемы, сохраняют антигенпрезентирующие свойства, также как и способность синтезировать множество цитокинов. Взаимодействие макрофаг / T-клетка зависит от присутствия множества костимулирующих молекул, включая членов семейства B7 (CD80 и CD86), некоторых молекул суперсемейства рецептора ФНО (CD40 и CD27) и CD5 колиганда CD72. Паттерн экспрессии CD80 и CD86 у легочных макрофагов пациентов с саркоидозом совместим с паттерном у нормальных APCs [17, 54, 55]. Действительно, саркоидные макрофаги увеличивают экспрессию молекул, которые наделяют макрофаги дополнительными функциями для активации и пролиферации T-клеток, включая CD80 и CD86, CD40 и CD72. Кроме того, было показано, что зрелые дендритные клетки могут быть обнаружены в центральном ядре саркоидных гранулем, предполагая, что субпопуляции дендритных клеток могут мигрировать из крови в пораженные ткани, способствуя формированию гранулемы [56].
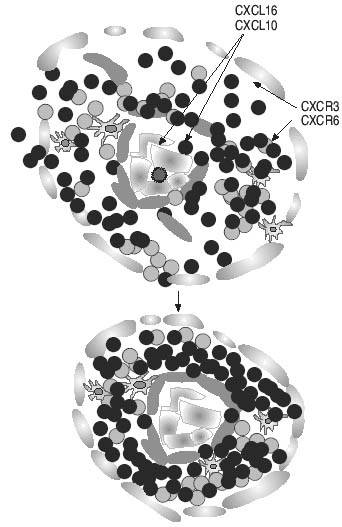
Недавно была исследована экспрессия генов цитокинов, которая в конечном счете объясняет накопление иммунокомпетентных клеток внутри гранулемы. IL-1b, IFN-g, CXCL16 и CXCL10, и CCL20 и CCL5 были экспрессированы клетками внутри гранулемы [29, 57, 58], принимая во внимание, что клетки, содержащие мРНК ФНО-a, IL-1a, IL-6 и IL-2 были рассеяны и распределены беспорядочно. Эти результаты говорят, что развитие новой гранулемы происходит при участии молекул с хемотаксическими свойствами, которые действуют совместно, чтобы рекрутировать моноциты и лимфоциты в периваскулярные очаги воспаления (рис. 2). Вероятно, производство IFN-g CD4 клетками является необходимым для формирования гранулемы, так как стимулировать формирование гранулем у мышей, с разрушенным геном IFN-g оказалось невозможным [59]. Хемокины, индуцированные IFN-g, также были вовлечены в формирование структуры гранулемы. Действительно, иммуногистологический анализ ткани показал, что клетки, несущие CXCL10 и CXCL16 - главным образом эпителиоидные клетки и CD68+ макрофаги, расположенные внутри гранулематозных областей (рис. 2). Оба хемокина функционально активны. Действительно, взаимодействуя со специфическими рецепторами, экспрессированными на Th1 клетках (CXCR6 и CXCR3), эти два хемокина способны стимулировать миграцию и накопление саркоидных T-лимфоцитов, которые окружают центральное ядро гранулемы.
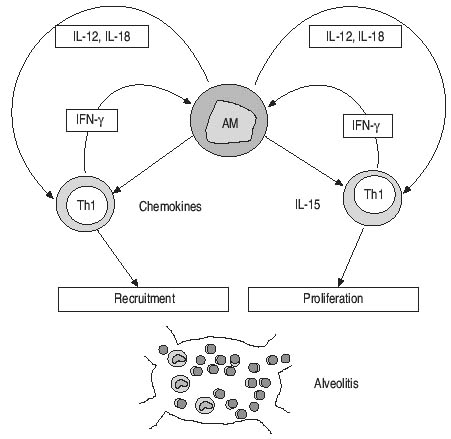
Рис. 2. Молекулы с хемотаксическими свойствами действуют совместно, чтобы рекрутировать моноциты в периваскулярные очаги саркоидного воспаления (см. секцию о хемокинах). Однако, клетки, несущие CXCL10 и CXCL16 расположены главным образом во внутренней части гранулемы. Действительно, эпителиоидные клетки и CD68+ макрофаги центрального ядра гранулемы в основном CXCL10+/CXCL16+, принимая во внимание, что лимфоциты, окружающие ядро гранулемы - CXCR3+/CXCR6+ T-клетки, предполагая, что взаимодействие между рецепторами хемокинов и их лигандами влияет на механизмы, ведущие формированию гранулемы.
Фиброз при саркоидозе
Так как при реакциях гиперчувствительности структура гранулемы нацелена на ограничение диссеминации агента, вызывающего реакцию, можно ожидать, что воспалительный ответ спонтанно разрешится, как только этиологический фактор будет изолирован. Эта парадигма не верна в случае рефрактерного саркоидоза. 60 % пациентов с саркоидозом имеют самоограниченный курс болезни, со спонтанным разрешением гранулем, принимая во внимание, что пациенты с прогрессирующим саркоидозом демонстрируют массовое развитие гранулем и ремиссия не происходит даже при проведении серьезной иммуносупрессивной терапии. Неконтролируемое развитие гранулем приводит к фиброзу.
Хотя медиаторами обратимой начальной стадии альвеолярного повреждения при саркоидозе являются Th1 лимфоциты, фиброзные изменения, которые следуют за саркоидным Th1 иммунным ответом, модулируются макрофагами, нейтрофилами, эозинофилами и тучными клетками [60, 61], которые посредством производства суперокисных анионов, свободных радикалов и протеаз, могут производить локальные повреждения и последующие нарушения нормальной архитектуры паренхимы легкого [62]. В подтверждение роли полиморфоядерных клеток в повреждении легкого, недавно было показано, что увеличенное число нейтрофилов в ЖБАЛ было связано с более серьезным курсом саркоидоза [63].
С помощью некоторых молекул, включая трансформирующий фактора роста бета (TGF-b) и семейство TGF цитокинов, фактор роста тромбоцитов и инсулин-подобный фактор роста I, саркоидные макрофаги, теоретически, могут быть медиаторами фиброза. Факторы роста фибробластов и эпителиоидных клеток и их рецепторов сильно экспрессированы в фиброзном легком. Вместе с семейством TGF они стимулируют рост фибробластов и накопление волокон коллагена. Кроме того, произведенные макрофагами цитокины (включая IL-1, IL-6, IFN-g, ФНО-a и GM-CSF) [64-66] и иммунные комплексы, на участках формирования гранулемы могут увеличивать экспрессию NO-синтазы и окиси азота [67], таким образом способствуя развитию повреждений и репаративных процессов.
Рекрутирование фибробластов и последующее увеличение производства матриксных макромолекул, являются важными для процесса фиброза. В частности, миграция фибробластов и эпителиоидных клеток из интерстиция к альвеолярным пространствам и адгезивные взаимодействия фибробластов с окружающим интерстициальным матриксом - главные факторы, способствующие развитию фиброза. Процесс миграции фибробластов отражает местное производство разнообразных молекул, таких как хемокины, продукты фибринолитического каскада и коагуляции, а также матриксные белки (коллагеновые пептиды, ламинин, фибронектин и эластин), которые могут действовать как хемоаттрактанты для фибробластов [68-71]. Большинство из них активно производится в легком при саркоидозе.
Молекулы, секретируемые саркоидными воспалительными клетками также способны стимулировать фибробласты входить в стадию ростового цикла G1 и, таким образом, пролиферировать. Действительно, большой процент фибробластов, изолированных у пациентов с легочным фиброзом, демонстрирует неожиданную способность к росту и более высокую частоту деления клеток, чем фибробласты, изолированные из нормального легкого. Кроме того, эти фибробласты имеют увеличенную миграционную способность [72].
Заключение
Начиная с 2000 г. было получено много новых данных относительно патогенеза саркоидоза. На основании на этих данных развиваются новые терапии. В настоящее время, основное внимание привлечено к растворимым цитокинам и ингибиторам хемокинов, генно-инженерным антагонистам, к комбинации нескольких или к отдельным противовоспалительным цитокинам, предполагая, что они могут стать стандартными фармакологическими средствами для контроля и модуляции иммунологических событий, которые ведут к формированию гранулемы и развитию фиброза. Например, молекулы, способные к нейтрализации ФНО-a использовались для лечения пациентов с саркоидозом [29, 73]. Эти данные предполагают реальную возможность использования анти-ФНО-a стратегии для лечения рефрактерного саркоидоза. Эти данные являются предварительными и в настоящее время проводятся клинические исследования, чтобы подтвердить эффективность и безопасность анти-ФНО-a терапии при лечении пациентов с саркоидозом. Ожидается, что блокада других воспалительных цитокинов, также будет полезна при саркоидозе и других T-клеточных диффузных болезнях легкого. В частности терапии, направленные на нейтрализацию хемокинов и других молекул, которые управляют траффиком и накоплением иммунокомпетентных клеток, потенциально более селективны и привлекательны, но требуют априорного знания о механизмах, регулирующих состояние воспаления альвеолярных и интерстициальных структур.
Резюме
Влияя на многие физиологические свойства иммунокомпетентных клеток, включая пролиферацию, дифференцирование, активацию и хемотаксис, хемокины и цитокины действуют как важные медиаторы функций клеток и взаимодействия клетка-клетка при саркоидозе. Фактически установлено, что в течение саркоидного воспалительного процесса, каскад внеклеточных сигнальных белков организует траффик иммунных клеток. Цитокины регулируют экспрессию молекул адгезии на сосудистом эндотелии легкого в пределах и вокруг участка воспаления, соединяясь с соответствующими рецепторами на соседних клетках. Это, в свою очередь, стимулирует рекрутирование и активацию Th1 клеток и локально модулирует процессы апоптоза и пролиферацию различных типов иммунных клеток, включая макрофаги, которые в свою очередь производят провоспалительные цитокины. В результате, цитокины с провоспалительными деструктивными биологическими функциями устанавливают состояние развития необратимого ремоделлирования ткани легкого, эволюцию к формированию легочных гранулем, и, у некоторых индивидуумов с саркоидозом, развитие фиброза.
Ключевые слова: цитокины, иммунопатогенез, саркоидоз, Т-леточный альвеолит.